Publications from the Hodges lab
2025
β-catenin functions as a molecular adapter for disordered cBAF interactions
BAF (SWI/SNF) chromatin remodelers engage binding partners to generate site-specific DNA accessibility. However, the basis for interaction between BAF and divergent binding partners has remained unclear. Here, we tested the hypothesis that scaffold proteins augment BAF's binding repertoire by examining β-catenin (CTNNB1) and steroidogenic factor 1 (SF-1, NR5A1), a transcription factor central to steroid production in human cells. BAF inhibition rapidly opposed SF-1/β-catenin enhancer occupancy, impairing SF-1 target activation and SF-1/β-catenin autoregulation. These effects arise due to β-catenin's role as a molecular adapter between SF-1 and an intrinsically disordered region (IDR) of the canonical BAF (cBAF) subunit ARID1A. In contrast to exclusively IDR-driven mechanisms, adapter function is mediated by direct association of ARID1A with β-catenin's folded Armadillo repeats. β-catenin similarly linked cBAF to YAP1, SOX2, FOXO3, and CBP/p300, reflecting a general IDR-mediated mechanism for modular coordination between factors. Molecular visualization highlights β-catenin's adapter role for interaction of cBAF with binding partners.
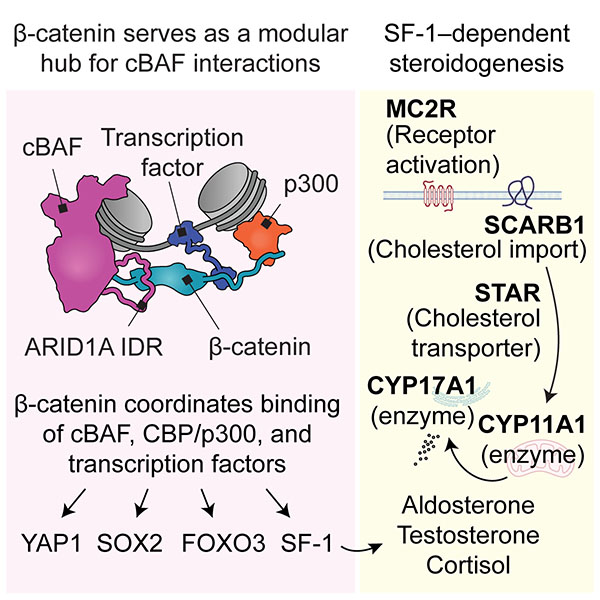
Chan YS, Gao Q, Robinson SA, Wang W, Filandrova R, Weinhold LM, Cabrera ML, Zhang M, Ambati CSR, Lerario AM, Putluri N, Kiseljak-Vassiliades K, Wierman ME, Habra MA, Hammer GD, Veverka V, Cermakova K, Hodges HC. β-catenin functions as a molecular adapter for disordered cBAF interactions. Mol Cell. 2025 Jul 15:S1097-2765(25)00576-3. doi: 10.1016/j.molcel.2025.06.026. PMID: 40695292.
IL-12-producing cytokine factories induce precursor exhausted T cells and elimination of primary and metastatic tumors
Curative responses to immunotherapy require the generation of robust systemic immunity with limited toxicity. Recruitment of T cell populations such as precursor exhausted T cells (Tpex) from lymphoid tissues to tumors is a hallmark of effective treatment. However, the ability to efficiently induce this recruitment is lacking in current immunotherapy approaches. Furthermore, systemic administration of immunotherapies frequently results in dose-limiting toxicities, yielding an inadequate therapeutic window for eliciting durable responses. In this investigation, we evaluated the safety and antitumor efficacy of locally administered interleukin 12 (IL-12) using a clinically translatable cytokine delivery platform (NCT05538624) to identify Tpex recruitment capabilities at tolerable cytokine doses. We show IL-12 cytokine factories can effectively treat a broad spectrum of cancer types. Single-cell RNA sequencing data suggests that the antitumor efficacy seen in our studies was due to retinal pigmented epithelial cells-mIL12 treatment inducing differentiation of Tpex cells within the tumor microenvironment. When administered in combination with checkpoint therapy, IL-12 cytokine factory treatment generated systemic abscopal immunity, preventing subcutaneous tumor outgrowth in 8/9 mice with colorectal cancer and lung metastasis in mice with melanoma. Furthermore, this platform was well tolerated in a non-human primate without signs of toxicity. Our new immunotherapy approach provides a robust strategy for inducing Tpex recruitment and systemic immunity against a range of solid peritoneal malignancies, many incurable with current immunotherapy strategies. Notably, these features were achieved using IL-12, and by leveraging our technology, we avoided the toxicities that have prevented the translation of IL-12 to the clinic. Our findings provide a strong rationale for the clinical development of IL-12 cytokine factories.
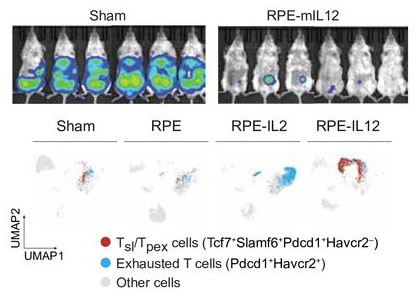
Nash A, DeBonis J, Murungi D, Castillo B, Kim B, Hu F, Chambers C, Nguyen A, Hernandez A, Wang Z, Rios PD, Ghani S, Joshi I, Isa D, Zheng N, Peng W, Igoshin OA, Oberholzer J, Hodges HC, Reticker-Flynn N, Veiseh O. IL-12-producing cytokine factories induce precursor exhausted T cells and elimination of primary and metastatic tumors. J Immunother Cancer. 2025 Apr 1;13(4):e010685. doi: 10.1136/jitc-2024-010685. PMID: 40169286; PMCID: PMC11962782.
2024
Pharmacologic Blockade of a Pioneer Transcription Factor
Cancers frequently co-opt lineage-specific transcription factors (TF) utilized in normal development to sustain proliferation. However, the effects of these TFs on tumor development depend considerably on where in the genome they bind. A new article by Taylor and colleagues expands on previously developed diamidine compounds that obstruct the DNA binding sites of the pioneer TF PU.1 (SPI1) in acute myeloid leukemia. Immobilization and sequencing of genomic DNA targeted by these compounds revealed that these inhibitors alter the genomic binding patterns of PU.1. The authors report that their strategy constrains the genomic binding preferences of PU.1, leading to redistribution of PU.1 to promoters and other gene-proximal regions with elevated guanine/cytosine content. Here, we discuss recent developments for targeting PU.1 in hematologic malignancies. We also explore the shared functional roles of PU.1 and SWI/SNF ATP-dependent chromatin remodeling complexes, which not only work together to sustain the enhancer landscape needed for tumor cell proliferation but also play key roles in nontumor settings. [PDF]
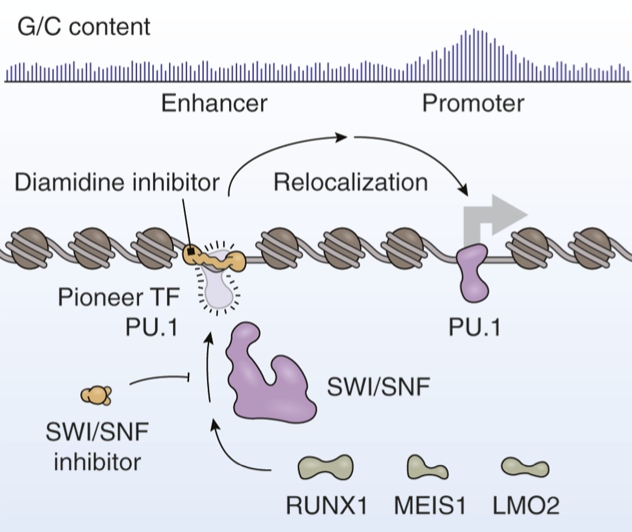
Cermakova K, Hodges HC. Pharmacologic Blockade of a Pioneer Transcription Factor. Cancer Res. 2024 Dec 16;84(24):4124-4125. doi: 10.1158/0008-5472.CAN-24-3957. PMID: 39476188.
Synergy of SWI/SNF inhibitors with retinoic acid: Reactivation of the G1 enhancer landscape underlies core circuitry addiction to SWI/SNF
Several cancer core regulatory circuitries (CRCs) depend on the sustained generation of DNA accessibility by SWI/SNF (BAF) chromatin remodelers. However, the window when SWI/SNF is acutely essential in these settings has not been identified. Here we used neuroblastoma (NB) cells to model and dissect the relationship between cell-cycle progression and SWI/SNF ATPase activity. We find that SWI/SNF inactivation impairs coordinated occupancy of non-pioneer CRC members at enhancers within 1 hour, rapidly breaking their autoregulation. By precisely timing inhibitor treatment following synchronization, we show that SWI/SNF is dispensable for survival in S and G2/M, but becomes acutely essential only during G1 phase. We furthermore developed a new approach to analyze the oscillating patterns of genome-wide DNA accessibility across the cell cycle, which revealed that SWI/SNF-dependent CRC binding sites are enriched at enhancers with peak accessibility during G1 phase, where they activate genes involved in cell-cycle progression. SWI/SNF inhibition strongly impairs G1-S transition and potentiates the ability of retinoids used clinically to induce cell-cycle exit. Similar cell-cycle effects in diverse SWI/SNF-addicted settings highlight G1-S transition as a common cause of SWI/SNF dependency. Our results illustrate that deeper knowledge of the temporal patterns of enhancer-related dependencies may aid the rational targeting of addicted cancers.
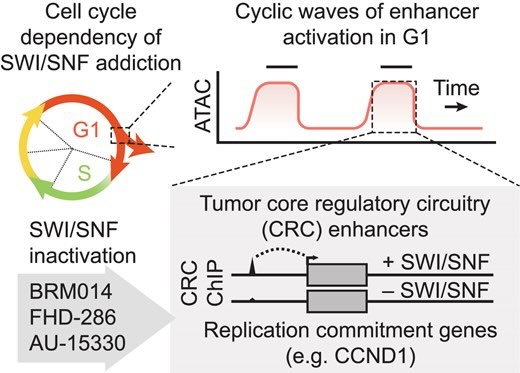
Cermakova K, Tao L, Dejmek M, Sala M, Montierth MD, Chan YS, Patel I, Chambers C, Loeza Cabrera M, Hoffman D, Parchem RJ, Wang W, Nencka R, Barbieri E, Hodges HC. Reactivation of the G1 enhancer landscape underlies core circuitry addiction to SWI/SNF. Nucleic Acids Res. 2024 Jan 11;52(1):4-21. doi: 10.1093/nar/gkad1081. PMID: 37993417; PMCID: PMC10783513.
The IL6/JAK/STAT3 signaling axis is a therapeutic vulnerability in SMARCB1-deficient bladder cancer
SMARCB1 loss has long been observed in many solid tumors. However, there is a need to elucidate targetable pathways driving growth and metastasis in SMARCB1-deficient tumors. Here, we demonstrate that SMARCB1 deficiency, defined as genomic SMARCB1 copy number loss associated with reduced mRNA, drives disease progression in patients with bladder cancer by engaging STAT3. SMARCB1 loss increases the chromatin accessibility of the STAT3 locus in vitro. Orthotopically implanted SMARCB1 knockout (KO) cell lines exhibit increased tumor growth and metastasis. SMARCB1-deficient tumors show an increased IL6/JAK/STAT3 signaling axis in in vivo models and patients. Furthermore, a pSTAT3 selective inhibitor, TTI-101, reduces tumor growth in SMARCB1 KO orthotopic cell line-derived xenografts and a SMARCB1-deficient patient derived xenograft model. We have identified a gene signature generated from SMARCB1 KO tumors that predicts SMARCB1 deficiency in patients. Overall, these findings support the clinical evaluation of STAT3 inhibitors for the treatment of SMARCB1-deficient bladder cancer.
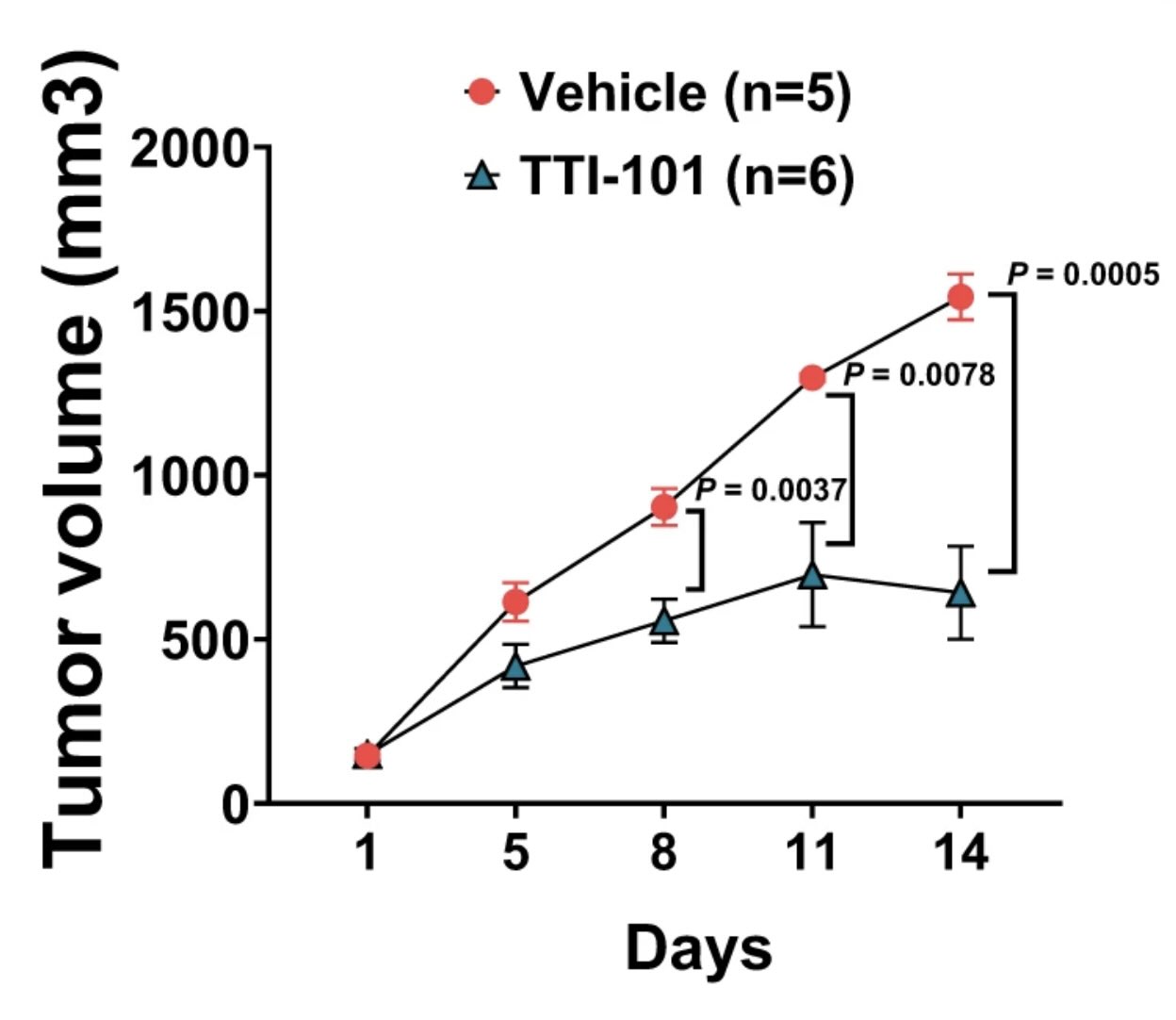
Amara CS, Kami Reddy KR, Yuntao Y, Chan YS, Piyarathna DWB, Dobrolecki LE, Shih DJH, Shi Z, Xu J, Huang S, Ellis MJ, Apolo AB, Ballester LY, Gao J, Hansel DE, Lotan Y, Hodges HC, Lerner SP, Creighton CJ, Sreekumar A, Zheng WJ, Msaouel P, Kavuri SM, Putluri N. The IL6/JAK/STAT3 signaling axis is a therapeutic vulnerability in SMARCB1-deficient bladder cancer. Nat Commun. 2024 Feb 14;15(1):1373. doi: 10.1038/s41467-024-45132-2. PMID: 38355560.
2023
Interaction modules that impart specificity to disordered protein
Intrinsically disordered regions (IDRs) are especially enriched among proteins that regulate chromatin and transcription. As a result, mechanisms that influence specificity of IDR-driven interactions have emerged as exciting unresolved issues for understanding gene regulation. We review the molecular elements frequently found within IDRs that confer regulatory specificity. In particular, we summarize the differing roles of disordered low-complexity regions (LCRs) and short linear motifs (SLiMs) towards selective nuclear regulation. Examination of IDR-driven interactions highlights SLiMs as organizers of selectivity, with widespread roles in gene regulation and integration of cellular signals. Analysis of recurrent interactions between SLiMs and folded domains suggests diverse avenues for SLiMs to influence phase-separated condensates and highlights opportunities to manipulate these interactions for control of biological activity.

Cermakova K, Hodges HC. Interaction modules that impart specificity to disordered protein. Trends Biochem Sci. 2023 May;48(5):477-490. doi: 10.1016/j.tibs.2023.01.004. Epub 2023 Feb 6. PMID: 36754681; PMCID: PMC10106370.
SWI/SNF blockade disrupts pioneer-directed enhancer programs in normal hematopoietic cells and acute myeloid leukemia
In acute myeloid leukemia (AML), SWI/SNF chromatin remodeling complexes sustain leukemic identity by driving high levels of MYC. Previous studies have implicated the hematopoietic transcription factor PU.1 (SPI1) as an important target of SWI/SNF inhibition, but PU.1 is widely regarded to have pioneer-like activity. As a result, many questions have remained regarding the interplay between PU.1 and SWI/SNF in AML as well as normal hematopoiesis. Here we found that PU.1 binds to most of its targets in a SWI/SNF-independent manner and recruits SWI/SNF to promote accessibility for other AML core regulatory factors, including RUNX1, LMO2, and MEIS1. SWI/SNF inhibition in AML cells reduced DNA accessibility and binding of these factors at PU.1 sites and redistributed PU.1 to promoters. Analysis of non-tumor hematopoietic cells revealed that similar effects also impair PU.1-dependent B cell and monocyte populations. Nevertheless, SWI/SNF inhibition induced profound therapeutic response in an immunocompetent AML mouse model as well as in primary human AML samples. In vivo, SWI/SNF inhibition promoted leukemic differentiation and reduced the leukemic stem cell burden in bone marrow but also induced leukopenia. These results reveal a variable therapeutic window for SWI/SNF blockade in AML and highlight important off-tumor effects of such therapies in immunocompetent settings.
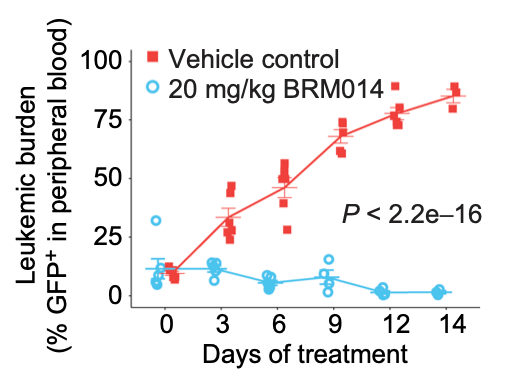
Chambers C, Cermakova K, Chan YS, Kurtz K, Wohlan K, Lewis AH, Wang C, Pham A, Dejmek M, Sala M, Loeza Cabrera M, Aguilar R, Nencka R, Lacorazza D, Rau RE, Hodges HC. SWI/SNF blockade disrupts PU.1-directed enhancer programs in normal hematopoietic cells and acute myeloid leukemia. Cancer Res. 2023 Jan 20:CAN-22-2129. doi: 10.1158/0008-5472.CAN-22-2129. Epub ahead of print. PMID: 36662812.
The TFIIS N-terminal domain (TND): a transcription assembly module at the interface of order and disorder
Interaction scaffolds that selectively recognize disordered protein strongly shape protein interactomes. An important scaffold of this type that contributes to transcription is the TFIIS N-terminal domain (TND). The TND is a five-helical bundle that has no known enzymatic activity, but instead selectively reads intrinsically disordered sequences of other proteins. Here, we review the structural and functional properties of TNDs and their cognate disordered ligands known as TND-interacting motifs (TIMs). TNDs or TIMs are found in prominent members of the transcription machinery, including TFIIS, super elongation complex, SWI/SNF, Mediator, IWS1, SPT6, PP1-PNUTS phosphatase, elongin, H3K36me3 readers, the transcription factor MYC, and others. We also review how the TND interactome contributes to the regulation of transcription. Because the TND is the most significantly enriched fold among transcription elongation regulators, TND- and TIM-driven interactions have widespread roles in the regulation of many transcriptional processes.
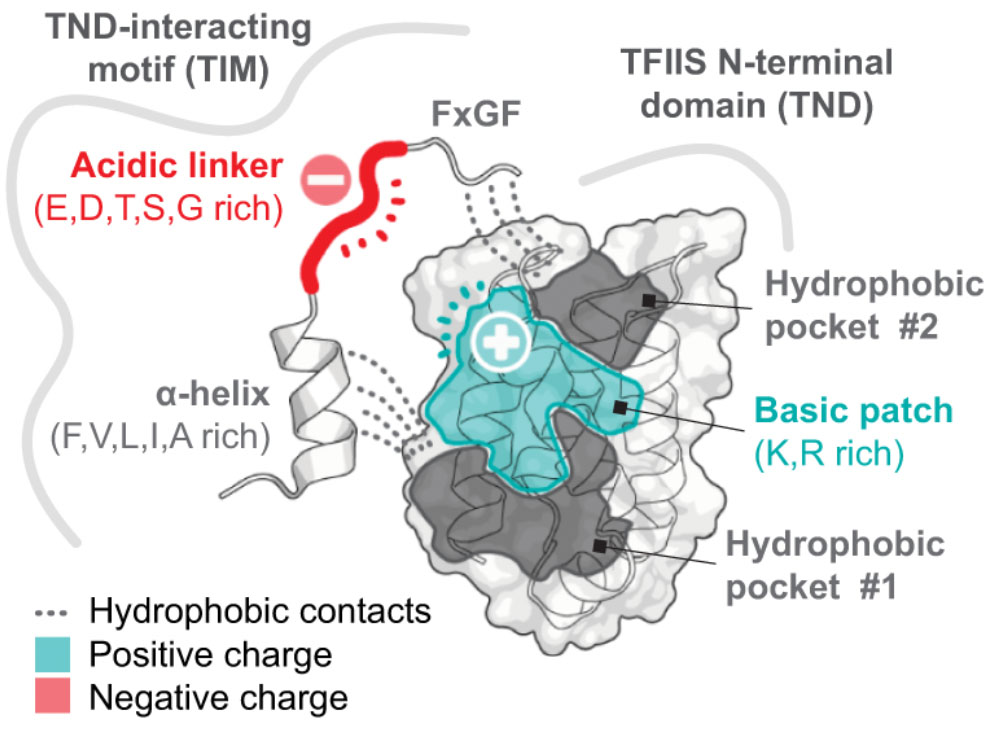
Cermakova K, Veverka V, Hodges HC. The TFIIS N-terminal domain (TND): a transcription assembly module at the interface of order and disorder. Biochem Soc Trans. 2023 Jan 18:BST20220342. doi: 10.1042/BST20220342. Epub ahead of print. PMID: 36651856.
2022
Immune profiling of adeno-associated virus response identifies B cell-specific targets that enable vector re-administration in mice.
Adeno-associated virus (AAV) vector-based gene therapies can be applied to a wide range of diseases. AAV expression can last for months to years, but vector re-administration may be necessary to achieve life-long treatment. Unfortunately, immune responses against these vectors are potentiated after the first administration, preventing the clinical use of repeated administration of AAVs. Reducing the immune response against AAVs while minimizing broad immunosuppression would improve gene delivery efficiency and long-term safety. In this study, we quantified the contributions of multiple immune system components of the anti-AAV response in mice. We identified B-cell-mediated immunity as a critical component preventing vector re-administration. Additionally, we found that IgG depletion alone was insufficient to enable re-administration, suggesting IgM antibodies play an important role in the immune response against AAV. Further, we found that AAV-mediated transduction is improved in µMT mice that lack functional IgM heavy chains and cannot form mature B-cells relative to wild-type mice. Combined, our results suggest that B-cells, including non-class switched B-cells, are a potential target for therapeutics enabling AAV re-administration. Our results also suggest that the µMT mice are a potentially useful experimental model for gene delivery studies since they allow repeated dosing for more efficient gene delivery from AAVs.
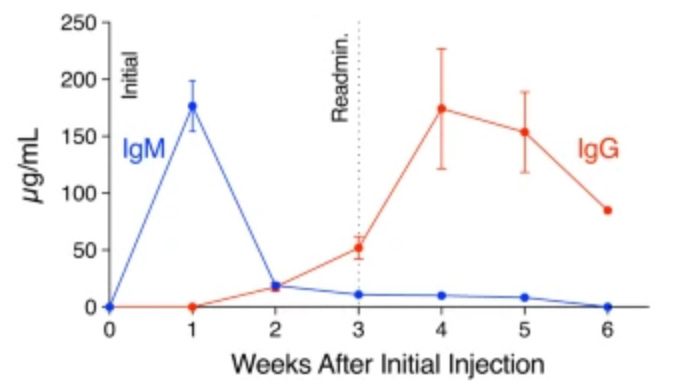
Chen M, Kim B, Jarvis MI, Fleury S, Deng S, Nouraein S, Butler S, Lee S, Chambers C, Hodges HC, Szablowski JO, Suh J, Veiseh O. Immune profiling of adeno-associated virus response identifies B cell-specific targets that enable vector re-administration in mice. Gene Ther. 2022 Nov 14. doi: 10.1038/s41434-022-00371-0. Epub ahead of print. PMID: 36372846.
2021
A ubiquitous disordered protein interaction module orchestrates transcription elongation
During eukaryotic transcription elongation, RNA polymerase II (RNAP2) is regulated by a chorus of factors. Here, we identified a common binary interaction module consisting of TFIIS N-terminal domains (TNDs) and natively unstructured TND-interacting motifs (TIMs). This module was conserved among the elongation machinery and linked complexes including transcription factor TFIIS, Mediator, super elongation complex, elongin, IWS1, SPT6, PP1-PNUTS phosphatase, H3K36me3 readers, and other factors. Using nuclear magnetic resonance, live-cell microscopy, and mass spectrometry, we revealed the structural basis for these interactions and found that TND-TIM sequences were necessary and sufficient to induce strong and specific colocalization in the crowded nuclear environment. Disruption of a single TIM in IWS1 induced robust changes in gene expression and RNAP2 elongation dynamics, which underscores the functional importance of TND-TIM surfaces for transcription elongation.
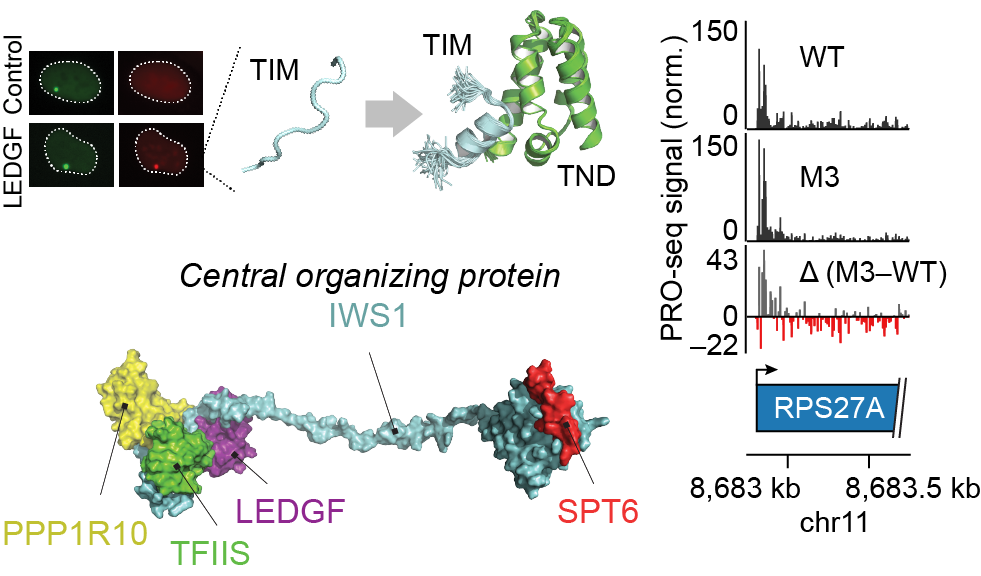
Cermakova K, Demeulemeester J, Lux V, Nedomova M, Goldman SR, Smith EA, Srb P, Hexnerova R, Fabry M, Madlikova M, Horejsi M, De Rijck J, Debyser Z, Adelman K, Hodges* HC, Veverka* V. A ubiquitous disordered protein interaction module orchestrates transcription elongation. Science. 2021 Nov 26;374(6571):1113-1121. doi: 10.1126/science.abe2913. Epub 2021 Nov 25. PMID: 34822292.
* Corresponding author.
The surface roughness of implanted prosthetics shapes immune response
Silicone is widely used in chronic implants and is generally perceived to be safe. However, textured breast implants have been associated with immune-related complications, including malignancies. Here, by examining for up to one year the foreign body response and capsular fibrosis triggered by miniaturized or full-scale clinically approved breast implants with different surface topography (average roughness, 0-90 μm) placed in the mammary fat pads of mice or rabbits, respectively, we show that surface topography mediates immune responses to the implants. We also show that the surface surrounding human breast implants collected during revision surgeries also differentially alters the individual's immune responses to the implant. Moreover, miniaturized implants with an average roughness of 4 μm can largely suppress the foreign body response and fibrosis (but not in T-cell-deficient mice), and that tissue surrounding these implants displayed higher levels of immunosuppressive FOXP3+ regulatory T cells. Our findings suggest that, amongst the topographies investigated, implants with an average roughness of 4 μm provoke the least amount of inflammation and foreign body response.
Doloff JC, Veiseh O, de Mezerville R, Sforza M, Perry TA, Haupt J, Jamiel M, Chambers C, Nash A, Aghlara-Fotovat S, Stelzel JL, Bauer SJ, Neshat SY, Hancock J, Romero NA, Hidalgo YE, Leiva IM, Munhoz AM, Bayat A, Kinney BM, Hodges HC, Miranda RN, Clemens MW, Langer R. The surface topography of silicone breast implants mediates the foreign body response in mice, rabbits and humans. Nat Biomed Eng. 2021 Jun 21. doi: 10.1038/s41551-021-00739-4. PMID: 34155355.
Non-canonical roles for PBRM1 during mitosis
Epigenetic effectors read marks written on chromatin to regulate function and fidelity of the genome. Here, we show that this coordinated read-write activity of the epigenetic machinery extends to the cytoskeleton, with PBRM1 in the PBAF chromatin remodeling complex reading microtubule methyl marks written by the SETD2 histone methyltransferase. PBRM1 binds SETD2 methyl marks via BAH domains, recruiting PBAF components to the mitotic spindle. This read-write activity was required for normal mitosis: Loss of SETD2 methylation or pathogenic BAH domain mutations disrupt PBRM1 microtubule binding and PBAF recruitment and cause genomic instability. These data reveal PBRM1 functions beyond chromatin remodeling with domains that allow it to integrate chromatin and cytoskeletal activity via its acetyl-binding BD and methyl-binding BAH domains, respectively. Conserved coordinated activity of the epigenetic machinery on the cytoskeleton opens a previously unknown window into how chromatin remodeler defects can drive disease via both epigenetic and cytoskeletal dysfunction.
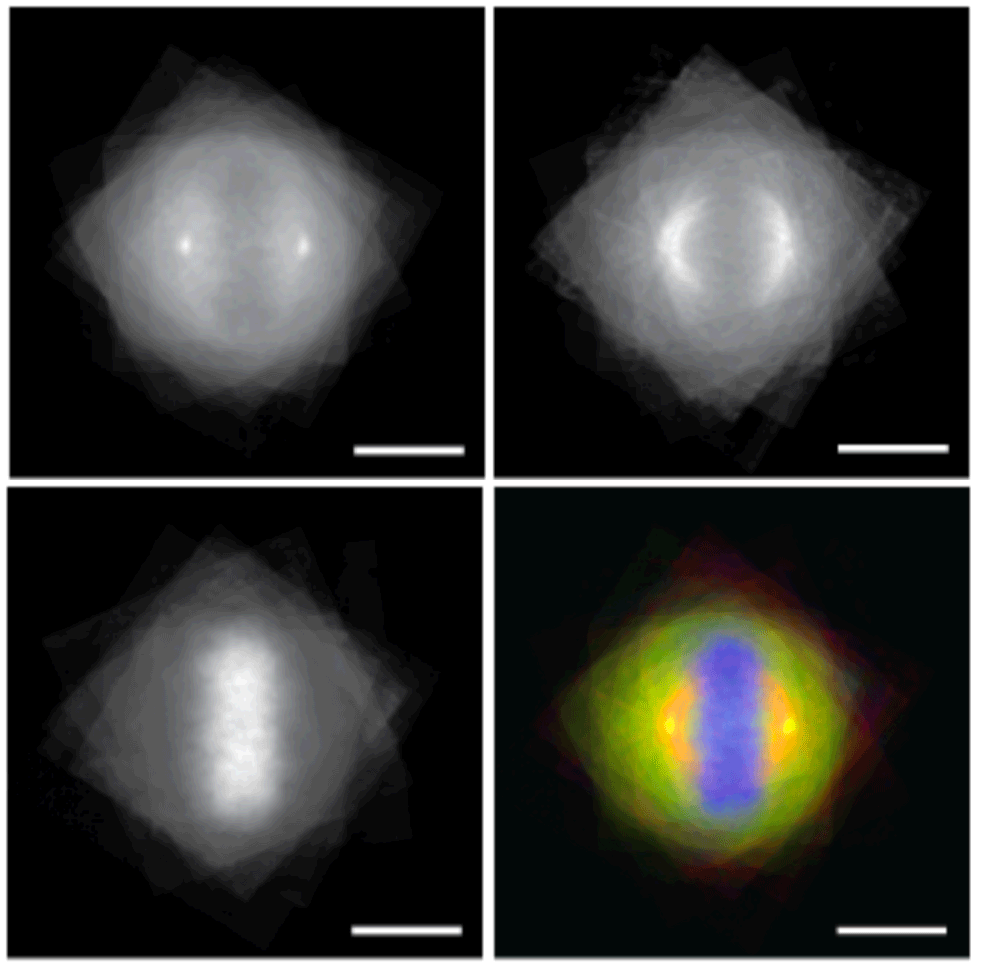
Karki M, Jangid RK, Anish R, Seervai RNH, Bertocchio JP, Hotta T, Msaouel P, Jung SY, Grimm SL, Coarfa C, Weissman BE, Ohi R, Verhey KJ, Hodges HC, Burggren W, Dere R, Park IY, Prasad BVV, Rathmell WK, Walker CL, Tripathi DN. A cytoskeletal function for PBRM1 reading methylated microtubules. Sci Adv. 2021 Apr 2;7(14):eabf2866. PMID: 33811077.
RELA oncofusion proteins in ependymoma
Over 60% of supratentorial (ST) ependymomas harbor a ZFTA-RELA (ZRfus) gene fusion (formerly C11orf95-RELA). To study the biology of ZRfus, we developed an autochthonous mouse tumor model using in utero electroporation (IUE) of the embryonic mouse brain. Integrative epigenomic and transcriptomic mapping was performed on IUE driven ZRfus tumors by CUT&RUN, ChIP, ATAC, and RNA sequencing and compared to human ZRfus driven ependymoma. In addition to direct canonical NF-kB pathway activation, ZRfus dictates a neoplastic transcriptional program and binds to thousands of unique sites across the genome that are enriched with Plagl family transcription factor (TF) motifs. ZRfus activates gene expression programs through recruitment of transcriptional co-activators (Brd4, Ep300, Cbp, Pol2) that are amenable to pharmacologic inhibition. Downstream ZRfus target genes converge on developmental programs marked by Plagl transcription factor proteins, and activate neoplastic programs enriched in Mapk, focal adhesion, and gene imprinting networks.
Arabzade A, Zhao Y, Varadharajan S, Chen HC, Jessa S, Rivas B, Stuckert AJ, Solis M, Kardian A, Tlais D, Golbourn BJ, Stanton AJ, Chan YS, Olson C, Karlin KL, Kong K, Kupp R, Hu B, Injac SG, Ngo M, Wang PR, De Leon LA, Sahm F, Kawauchi D, Pfister SM, Lin CY, Hodges HC, Singh I, Westbrook TF, Chintagumpala MM, Blaney SM, Parsons DW, Pajtler KW, Agnihotri S, Gilbertson RJ, Yi J, Jabado N, Kleinman CL, Bertrand KC, Deneen B, Mack SC. ZFTA-RELA Dictates Oncogenic Transcriptional Programs to Drive Aggressive Supratentorial Ependymoma. Cancer Discov. 2021 Mar 19:candisc.1066.2020. PMID: 33741710.
2020
CHD8 in autism spectrum disorder and early neural development
The chromatin remodeler CHD8 is among the most frequently mutated genes in autism spectrum disorder (ASD). CHD8 has a dosage-sensitive role in ASD, but when and how it becomes critical to human social function is unclear. Here, we conducted genomic analyses of heterozygous and homozygous Chd8 mouse embryonic stem cells and differentiated neural progenitors. We identify dosage-sensitive CHD8 transcriptional targets, sites of regulated accessibility, and an unexpected cooperation with SOX transcription factors. Collectively, our findings reveal that CHD8 negatively regulates expression of neuronal genes to maintain pluripotency and also during differentiation. Thus, CHD8 is essential for both the maintenance of pluripotency and neural differentiation, providing mechanistic insight into its function with potential implications for ASD.
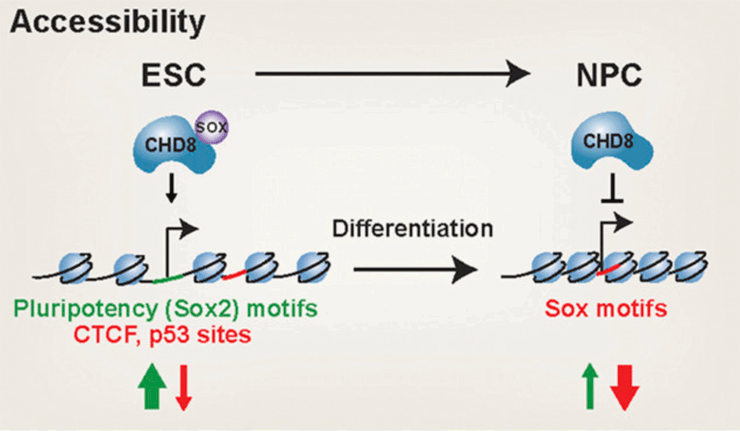
Sood S, Weber CM, Hodges HC, Krokhotin A, Shalizi A, Crabtree GR. CHD8 dosage regulates transcription in pluripotency and early murine neural differentiation. Proc Natl Acad Sci U S A. 2020 Sep 8;117(36):22331-22340. PMID: 32839322; PMCID: PMC7486765
2019
Genome-wide approaches for studying tumor heterogeneity
Many malignancies display heterogeneous features that support cancer progression. Emerging high-resolution methods provide a view of heterogeneity that recognizes the influence of diverse cell types and cell states of the tumor microenvironment. Here we outline a hierarchical organization of tumor heterogeneity from a genomic perspective, summarize the origins of spatially patterned metabolic features, and review recent developments in single-cell and spatially resolved techniques for genome-wide study of multicellular tissues. We also discuss how integrating these approaches can yield new insights into human cancer and emerging immune therapies. Applying these technologies for the analysis of primary tumors, patient-derived xenografts, and in vitro systems holds great promise for understanding the hierarchical structure and environmental influences that underlie tumor ecosystems. [PDF]
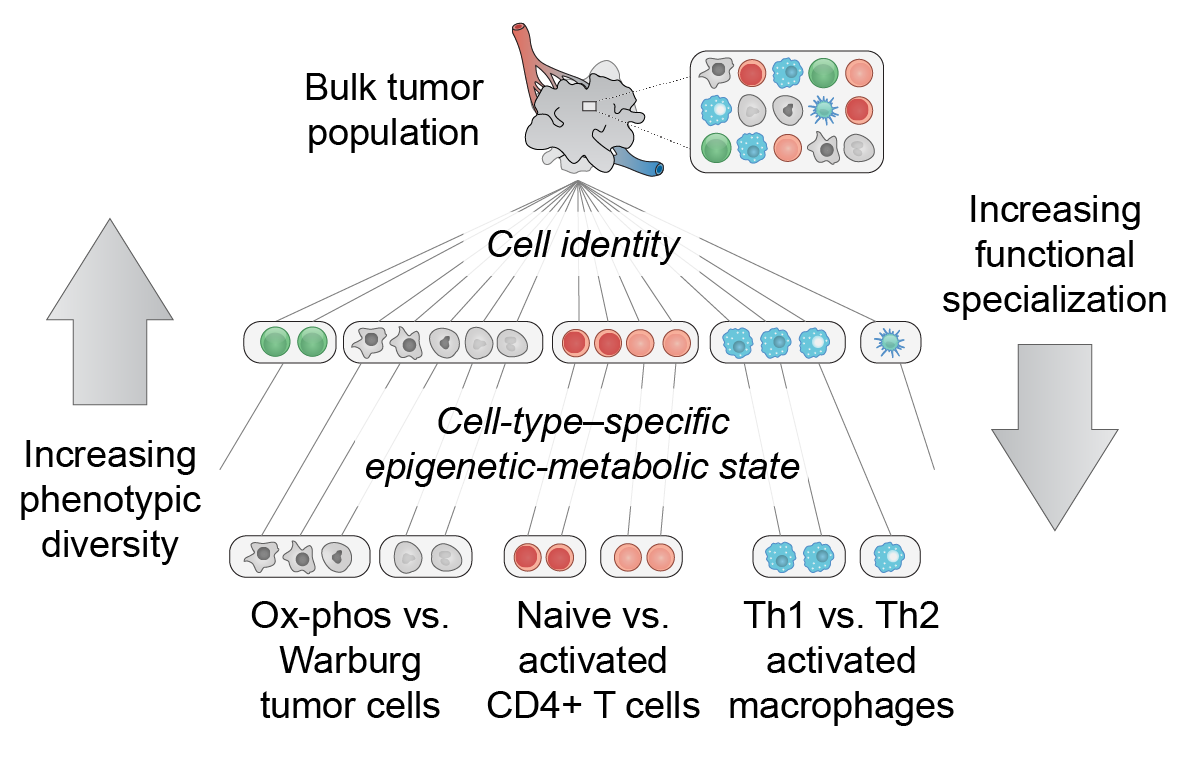
EA Smith & HC Hodges, "The spatial and genomic hierarchy of tumor ecosystems revealed by single-cell technologies" Trends in Cancer 5(7): 411-425 (2019).
cAMP-dependent allosteric regulation of Protein Kinase A (PKA)
Cyclic nucleotide binding (CNB) domains allosterically regulate the activity of proteins with diverse functions, but the mechanisms that enable the cyclic nucleotide binding signal to regulate distant domains are not well understood. Here we use optical tweezers and molecular dynamics to dissect changes in folding energy landscape associated with cAMP binding signals transduced between the two CNB domains of protein kinase A (PKA). We find that the response of the energy landscape upon cAMP binding is domain-specific, resulting in unique but mutually coordinated tasks: one CNB domain initiates cAMP binding and cooperativity, while the other triggers inter-domain interactions that promote the active conformation. Inter-domain interactions occur in a stepwise fashion, beginning in intermediate-liganded states between apo and cAMP-bound domains. Moreover, we identify a cAMP-responsive switch, the N3A motif, whose conformation and stability depend on cAMP occupancy. This switch serves as a signaling hub, amplifying cAMP binding signals during PKA activation.
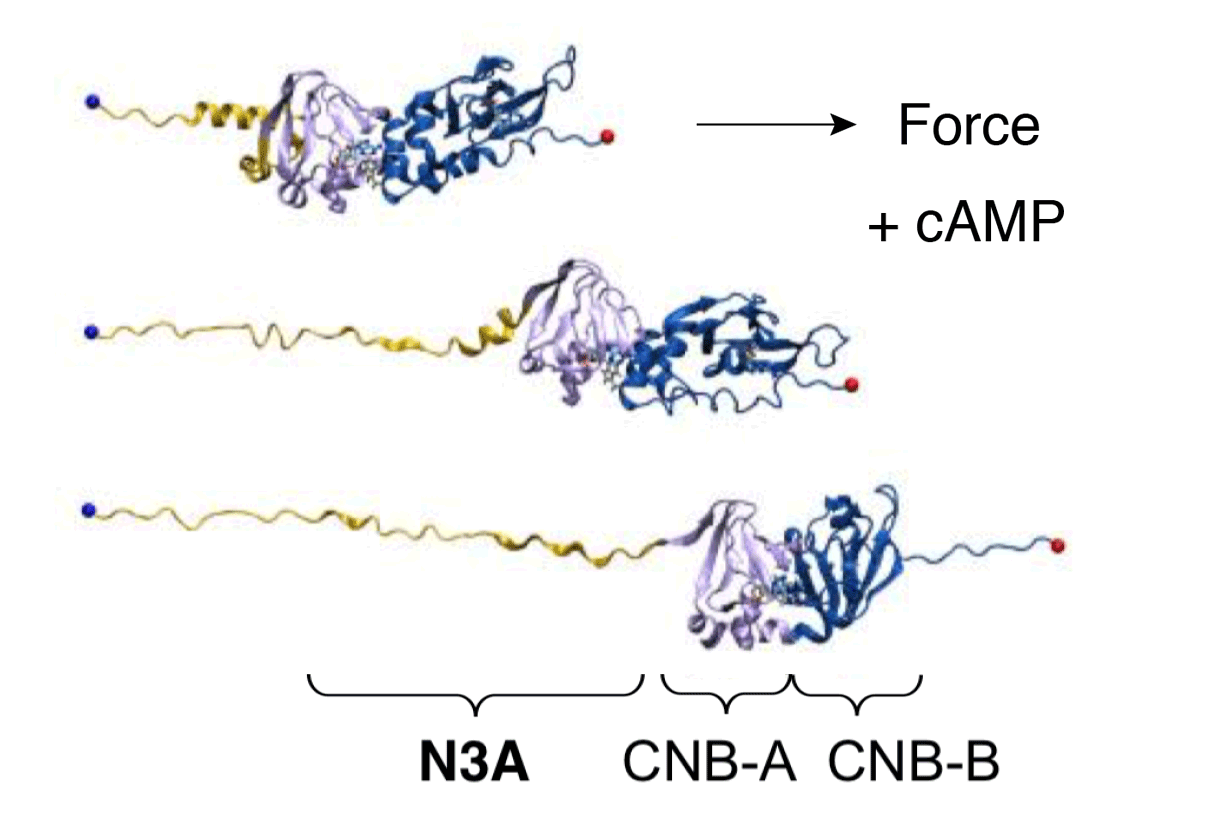
Y Hao, J England, L Bellucci, E Paci, HC Hodges, S Taylor, RA Maillard. "Activation of PKA via asymmetric allosteric coupling of structurally conserved cyclic nucleotide binding domains" Nature Communications 10(1): 3984, (2019).
Expert consensus guidelines for clinical management of a SMARCB1-deficient cancer
Renal medullary carcinoma (RMC) is defined in part by loss of SMARCB1, a subunit of BAF (SWI/SNF) and PBAF complexes. RMC is one of the most aggressive renal cell carcinomas. It predominantly afflicts young adults and adolescents with sickle cell trait and other sickle hemoglobinopathies, and is refractory to targeted and anti-angiogenic therapies used in patients with clear-cell renal cell carcinoma. Platinum-based cytotoxic chemotherapy is the mainstay for RMC treatment. Based on recent advances in the diagnosis, management, and clinical trial development for RMC, a panel of experts met in October 2017 and developed updated consensus recommendations to inform clinicians, researchers, and patients. Because RMC often aggressively recurs while patients are still recovering from nephrectomy, upfront chemotherapy should be considered for the majority of patients, including those with localized disease. After safety and dosing information have been established in adults, phase II and III trials enrolling patients with RMC should allow patients aged 12 years and older to be accrued. Patients with the very rare unclassified renal cell carcinoma with medullary phenotype variant should be included in RMC trials. Medical providers should be aware that RMC can afflict subjects of all races, and not only those of the African descent, and that the presence of sickle cell trait, or of other sickle hemoglobinopathies, can impact drug responses and toxicity.

P Msaouel, AL Hong, EA Mullen, MB Atkins, CL Walker, CH Lee, MA Carden, G Genovese, WM Linehan, P Rao, MJ Merino, H Grodman, JS Dome, CV Fernandez, JI Geller, AB Apolo, NC Daw, HC Hodges, M Moxey-Mims, D Wei, DP Bottaro, M Staehler, JA Karam, WK Rathmell, NM Tannir (2018) "Updated recommendations on the diagnosis, management, and clinical trial eligibility criteria for patients with renal medullary carcinoma" Clinical Genitourinary Cancer 17(1):1-6 (2019).
2018
Next-generation drugs and probes for chromatin biology: From targeted protein degradation to phase separation
Chromatin regulation is a critical aspect of nuclear function. Mechanisms crucial for normal nuclear function and epigenetic control include compartmentalization of biochemical reactions by liquid-phase separated condensates and signal-dependent regulation of protein stability. Synthetic control of these phenomena by small molecules provides deep insight into essential activities such as histone modification, BAF (SWI/SNF) and PBAF remodeling, Polycomb repression, enhancer looping by cohesin and CTCF, as well as many other processes that contribute to transcription. As a result, a complete understanding of the spatiotemporal mechanisms that underlie chromatin regulation increasingly requires the use of fast-acting drugs and chemical probes. Here, we provide a comprehensive review of next-generation chemical biology tools to interrogate the chromatin regulatory landscape, including selective PROTAC E3 ubiquitin ligase degraders, degrons, fluorescent ligands, dimerizers, inhibitors, and other drugs. These small molecules provide important insights into the mechanisms that govern gene regulation, DNA repair, development, and diseases like cancer.
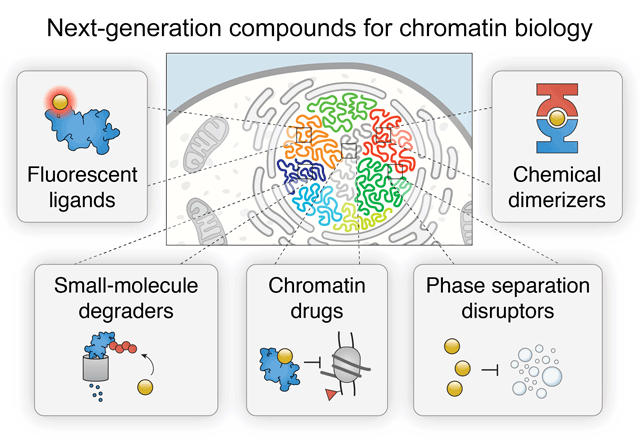
Cermakova K, Hodges HC (2018) "Next-Generation Drugs and Probes for Chromatin Biology: From Targeted Protein Degradation to Phase Separation" Molecules 23(8): 1958.
New principles behind cAMP-regulated allostery and multidomain folding
Protein kinases are dynamic molecular switches that sample multiple conformational states. The regulatory subunit of protein kinase A (PKA) harbors two cAMP-binding domains [cyclic nucleotide-binding (CNB) domains] that oscillate between inactive and active conformations dependent on cAMP binding. The cooperative binding of cAMP to the CNB domains activates an allosteric interaction network that enables PKA to progress from the inactive to active conformation, unleashing the activity of the catalytic subunit. Despite its importance in the regulation of many biological processes, the molecular mechanism responsible for the observed cooperativity during the activation of PKA remains unclear. Here, we use optical tweezers to probe the folding cooperativity and energetics of domain communication between the cAMP-binding domains in the apo state and bound to the catalytic subunit. Our study provides direct evidence of a switch in the folding-energy landscape of the two CNB domains from energetically independent in the apo state to highly cooperative and energetically coupled in the presence of the catalytic subunit. Moreover, we show that destabilizing mutational effects in one CNB domain efficiently propagate to the other and decrease the folding cooperativity between them. Taken together, our results provide a thermodynamic foundation for the conformational plasticity that enables protein kinases to adapt and respond to signaling molecules.
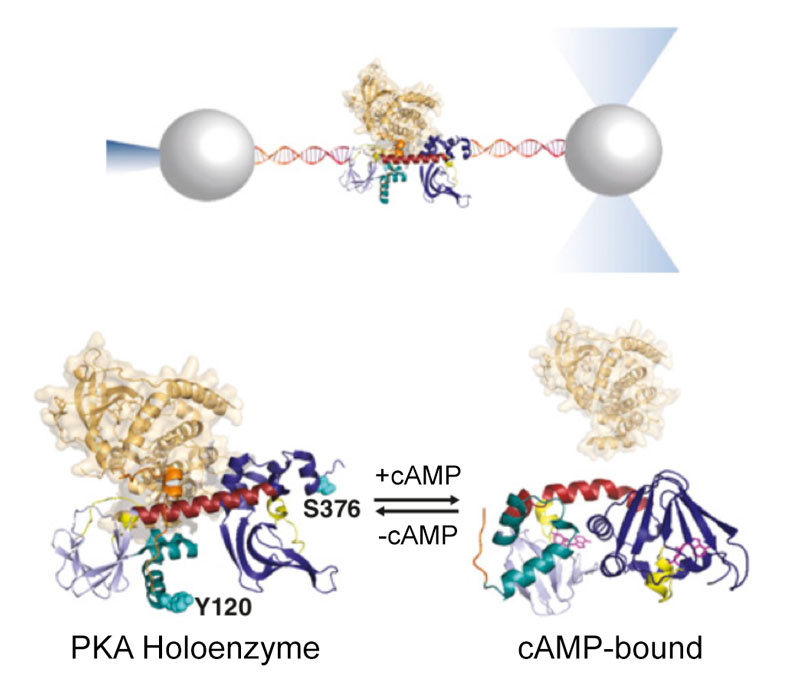
England JP, Hao Y, Bai L, Glick V, Hodges HC, Taylor SS and Maillard RA (2018) "Switching of the folding-energy landscape governs the allosteric activation of protein kinase A" Proc Natl Acad Sci USA 115(32): 7478-7485.
Kinase-regulated interactions with LEDGF (PSIP1), an H3K36me3 reader
Lens epithelium-derived growth factor/p75 (LEDGF/p75, or PSIP1) is a transcriptional coactivator that tethers other proteins to gene bodies. The chromatin tethering function of LEDGF/p75 is hijacked by HIV integrase to ensure viral integration at sites of active transcription. LEDGF/p75 is also important for the development of mixed-lineage leukemia (MLL), where it tethers the MLL1 fusion complex at aberrant MLL targets, inducing malignant transformation. However, little is known about how the LEDGF/p75 protein interaction network is regulated. Here, we obtained solution structures of the complete interfaces between the LEDGF/p75 integrase binding domain (IBD) and its cellular binding partners and validated another binding partner, Mediator subunit 1 (MED1). We reveal that structurally conserved IBD-binding motifs (IBMs) on known LEDGF/p75 binding partners can be regulated by phosphorylation, permitting switching between low- and high-affinity states. Finally, we show that elimination of IBM phosphorylation sites on MLL1 disrupts the oncogenic potential of primary MLL1-rearranged leukemic cells. Our results demonstrate that kinase-dependent phosphorylation of MLL1 represents a previously unknown oncogenic dependency that may be harnessed in the treatment of MLL-rearranged leukemia.
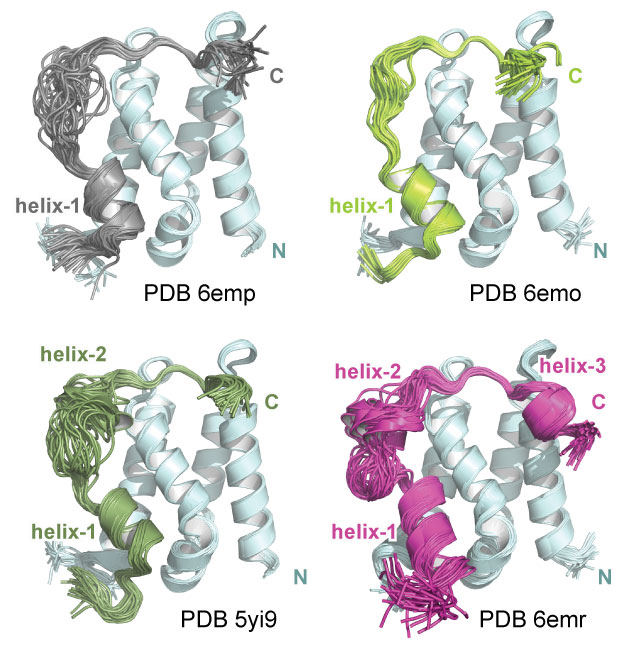
Sharma S*, Cermakova K*, De Rijck J, Demeulemeester J, Fabry M, El Ashkar S, Van Belle S, Lepsik M, Tesina P, Duchoslav V, Novak P, Hubalek M, Srb P, Christ F, Rezacova P, Hodges HC, Debyser Z, Veverka V (2018) "Affinity switching of the LEDGF/p75 IBD interactome is governed by kinase-dependent phosphorylation" Proc Natl Acad Sci USA 115(30): 7053-7062.
Enhancers disrupted by dominant-negative SMARCA4 (BRG1) mutations
Mutation of SMARCA4 (BRG1), the ATPase of BAF (mSWI/SNF) and PBAF complexes, contributes to a range of malignancies and neurologic disorders. Unfortunately, the effects of SMARCA4 missense mutations have remained uncertain. Here we show that SMARCA4 cancer missense mutations target conserved ATPase surfaces and disrupt the mechanochemical cycle of remodeling. We find that heterozygous expression of mutants alters the open chromatin landscape at thousands of sites across the genome. Loss of DNA accessibility does not directly overlap with Polycomb accumulation, but is enriched in 'A compartments' at active enhancers, which lose H3K27ac but not H3K4me1. Affected positions include hundreds of sites identified as superenhancers in many tissues. Dominant-negative mutation induces pro-oncogenic expression changes, including increased expression of Myc and its target genes. Together, our data suggest that disruption of enhancer accessibility represents a key source of altered function in disorders with SMARCA4 mutations in a wide variety of tissues.
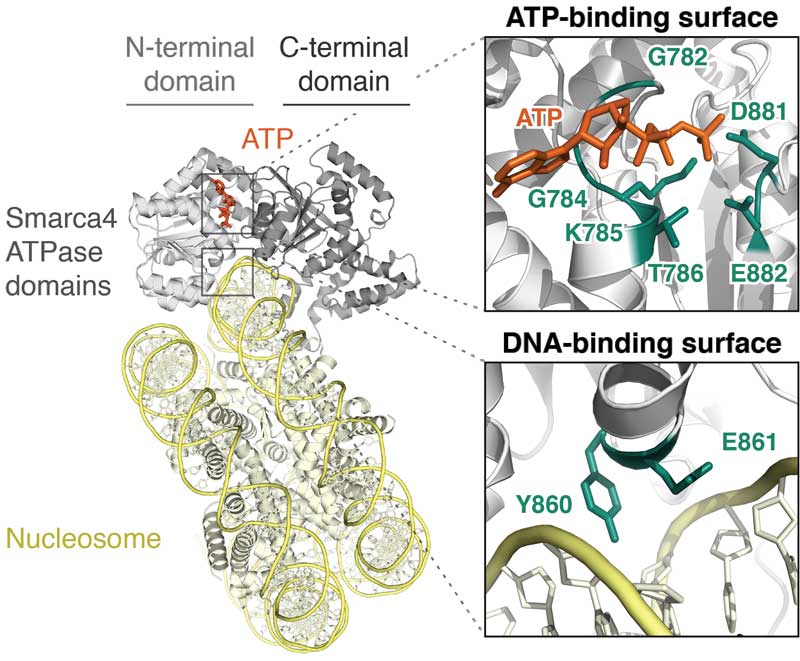
Hodges HC*, Stanton BZ*, Cermakova K, Chang CY, Miller EL, Kirkland JG, Ku WL, Veverka V, Zhao K, Crabtree GR (2018) "Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers," Nature Structural and Molecular Biology 25(1): 61-72.
2017
Synergy between chromatin remodeling and topoisomerase activity
The resolution and formation of facultative heterochromatin are essential for development, reprogramming, and oncogenesis. The mechanisms underlying these changes are poorly understood owing to the difficulty of studying heterochromatin dynamics and structure in vivo. We devised an in vivo approach to investigate these mechanisms and found that topoisomerase II (TOP2), but not TOP1, synergizes with BAF (mSWI/SNF) ATP-dependent chromatin remodeling complexes genome-wide to resolve facultative heterochromatin to accessible chromatin independent of transcription. This indicates that changes in DNA topology that take place through (de-)catenation rather than the release of torsional stress through swiveling are necessary for heterochromatin resolution. TOP2 and BAF cooperate to recruit pluripotency factors, which explains some of the instructive roles of BAF complexes. Unexpectedly, we found that TOP2 also plays a role in the re-formation of facultative heterochromatin; this finding suggests that facultative heterochromatin and accessible chromatin exist at different states of catenation or other topologies, which might be critical to their structures.
Miller EL, Hargreaves DC, Kadoch C, Chang CY, Calarco JP, Hodges C, Buenrostro JD, Cui K, Greenleaf WJ, Zhao K, Crabtree GR (2017) "TOP2 synergizes with BAF chromatin remodeling for both resolution and formation of facultative heterochromatin," Nature Structural and Molecular Biology 24(4): 344-352.
Smarca4 (Brg1) directly regulates Polycomb repression by PRC1 at bivalent genes
Trithorax-group proteins and their mammalian homologs, including those in BAF (mSWI/SNF) complexes, are known to oppose the activity of Polycomb repressive complexes (PRCs). This opposition underlies the tumor-suppressive role of BAF subunits and is expected to contribute to neurodevelopmental disorders. However, the mechanisms underlying opposition to Polycomb silencing are poorly understood. Here we report that recurrent disease-associated mutations in BAF subunits induce genome-wide increases in PRC deposition and activity. We show that point mutations in SMARCA4 (also known as BRG1) mapping to the ATPase domain cause loss of direct binding between BAF and PRC1 that occurs independently of chromatin. Release of this direct interaction is ATP dependent, consistent with a transient eviction mechanism. Using a new chemical-induced proximity assay, we find that BAF directly evicts Polycomb factors within minutes of its occupancy, thereby establishing a new mechanism for the widespread BAF-PRC opposition underlying development and disease.
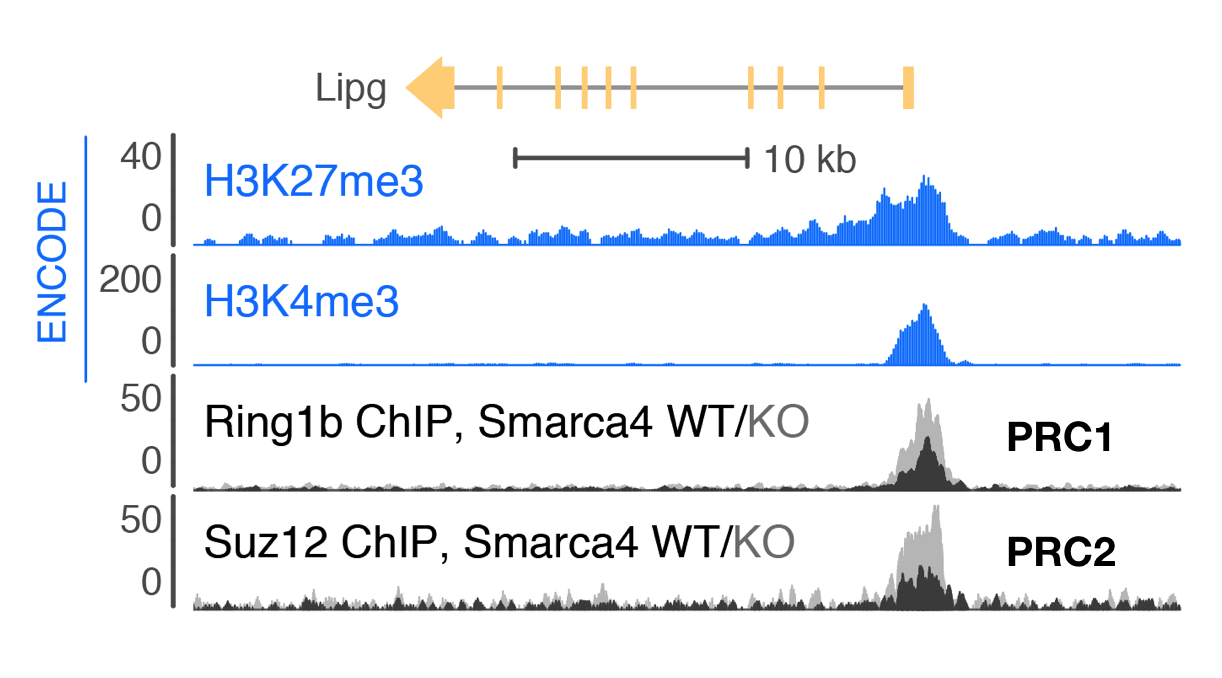
Stanton BZ*, Hodges C*, Calarco JP, Braun SM, Ku WL, Kadoch C, Zhao K, Crabtree GR (2017) "Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin," Nature Genetics 49(2): 282-288.
2016
Review: Subunit-specific roles of BAF (SWI/SNF) and PBAF complexes in cancer
During the last decade, a host of epigenetic mechanisms were found to contribute to cancer and other human diseases. Several genomic studies have revealed that ~20% of malignancies have alterations of the subunits of polymorphic BAF and PBAF complexes, making them among the most frequently mutated complexes in cancer. Recurrent mutations arise in genes encoding several BAF/PBAF subunits, including ARID1A, ARID2, PBRM1, SMARCA4, and SMARCB1. These subunits share some degree of conservation with subunits from related ATP-dependent chromatin remodeling complexes in model organisms, where a large body of work provides insight into their roles in cancer. Here we review the roles of BAF- and PBAF-like complexes in these organisms, and relate these findings to recent discoveries in cancer epigenomics. We review several roles of BAF and PBAF complexes in cancer, including transcriptional regulation, DNA repair, and regulation of chromatin architecture and topology. More recent results highlight the need for new techniques to study these complexes. [PDF]
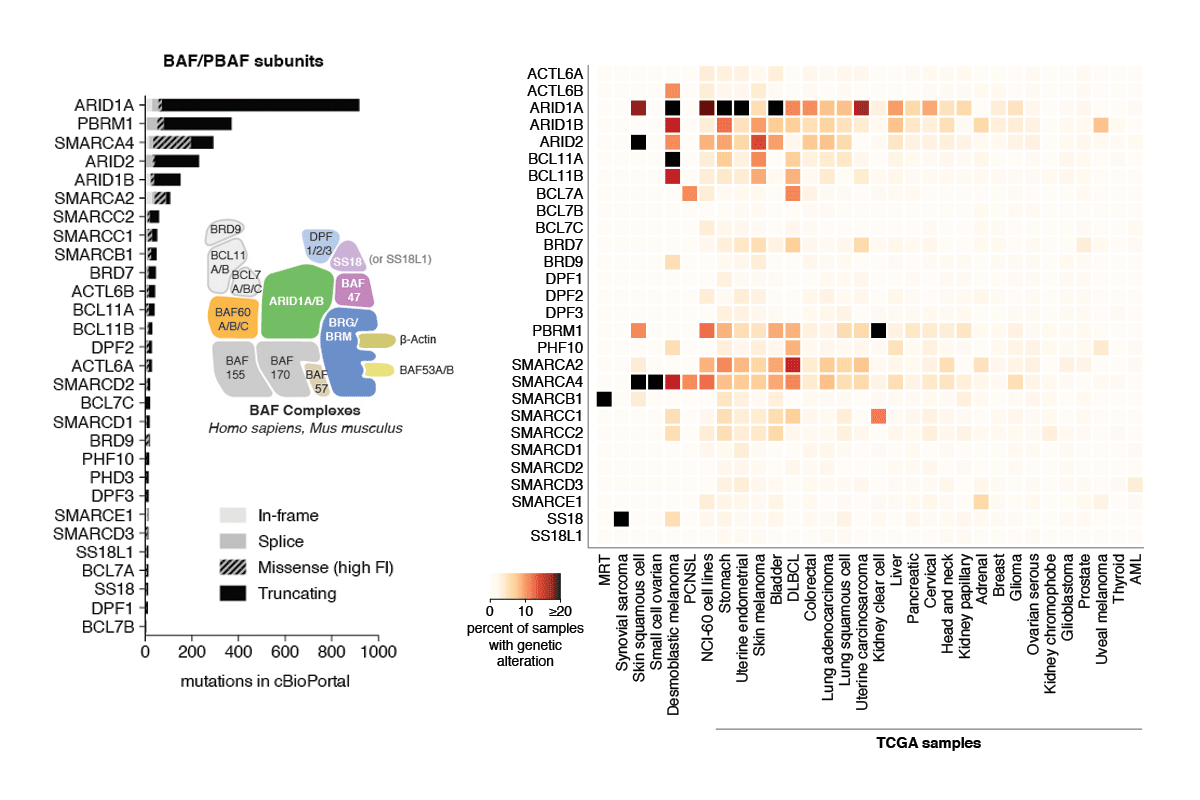
Hodges C, Kirkland JK, and Crabtree GR (2016) "The many roles of BAF (mSWI/SNF) and PBAF complexes in cancer," Cold Spring Harbor Perspectives in Medicine 6(8): pii: a026930.
2013
Meta-analysis of tumor exome sequencing studies reveals an extensive role of SWI/SNF in human malignancy
Subunits of mammalian SWI/SNF (mSWI/SNF or BAF) complexes have recently been implicated as tumor suppressors in human malignancies. To understand the full extent of their involvement, we conducted a proteomic analysis of endogenous mSWI/SNF complexes, which identified several new dedicated, stable subunits not found in yeast SWI/SNF complexes, including BCL7A, BCL7B and BCL7C, BCL11A and BCL11B, BRD9 and SS18. Incorporating these new members, we determined mSWI/SNF subunit mutation frequency in exome and whole-genome sequencing studies of primary human tumors. Notably, mSWI/SNF subunits are mutated in 19.6% of all human tumors reported in 44 studies. Our analysis suggests that specific subunits protect against cancer in specific tissues. In addition, mutations affecting more than one subunit, defined here as compound heterozygosity, are prevalent in certain cancers. Our studies demonstrate that mSWI/SNF is the most frequently mutated chromatin-regulatory complex (CRC) in human cancer, exhibiting a broad mutation pattern, similar to that of TP53. Thus, proper functioning of polymorphic BAF complexes may constitute a major mechanism of tumor suppression.

Kadoch C*, Hargreaves DC*, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR (2013). "Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy," Nature Genetics 45(6): 592-601.
2012
Computational modeling of histone modification domain spreading
A central goal of chromatin biology is to reveal how posttranslational histone marks modulate gene expression; however, relatively little is known about the spatial or temporal dynamics of these marks. We previously showed that a dynamic model of histone mark nucleation, propagation, and turnover fits the mean enrichment profiles from 99% of noncentromeric histone H3 lysine 9 trimethylation (H3K9me3) domains in mouse embryonic stem cells without the need for boundary or insulator elements. Here we report the full details of this "inherently bounded" model of histone modification dynamics and describe several dynamic features of the model using H3K9me3 as a paradigm. By analyzing the kinetic and structural constraints that drive formation of inherently bounded domains, we find that such domains are optimized when the rates of marking and turnover are comparable. Additionally, we find that to establish such domains, propagation of the histone marks must occur primarily through local contacts.
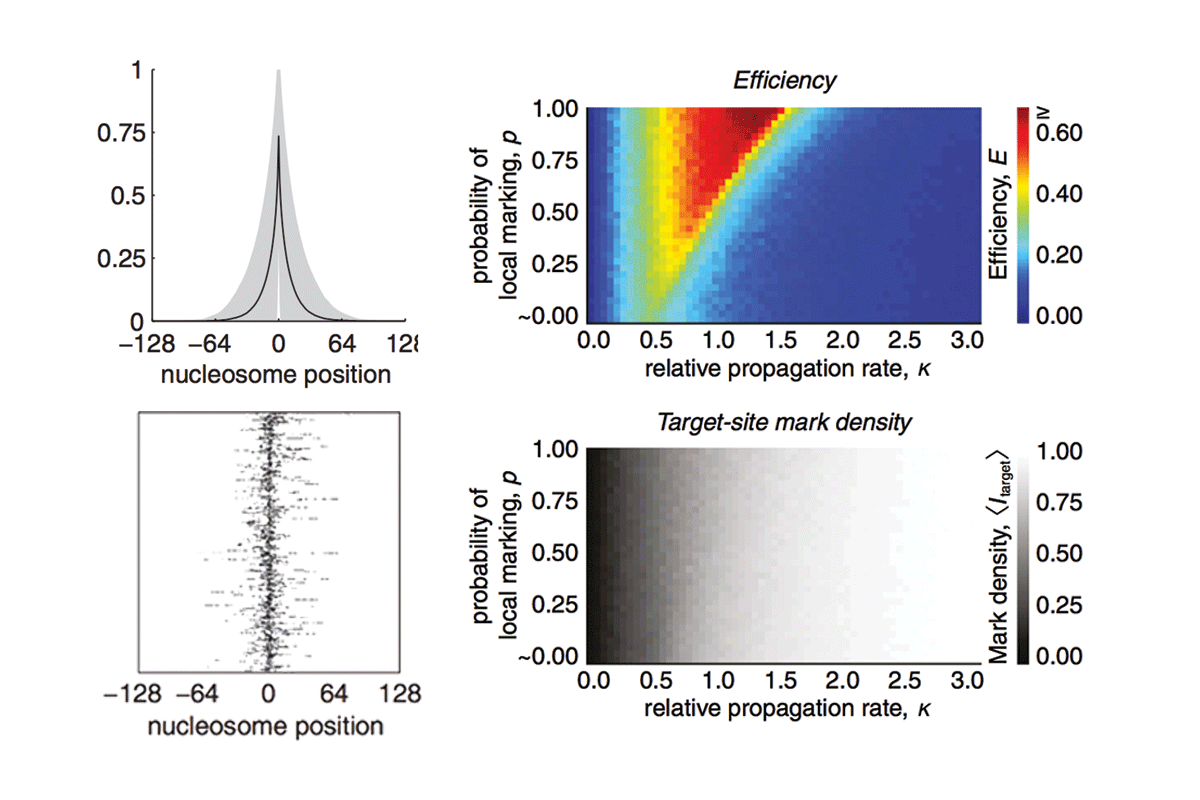
Hodges C and Crabtree GR (2012) "Dynamics of inherently bounded histone modification domains" PNAS 109(33): 13296-13301.
Rewriting the epigenome with small-molecule induced chromatin modifications
Posttranslational histone modifications are important for gene regulation, yet the mode of propagation and the contribution to heritable gene expression states remains controversial. To address these questions, we developed a chromatin in vivo assay (CiA) system employing chemically induced proximity to initiate and terminate chromatin modifications in living cells. We selectively recruited HP1α to induce H3K9me3-dependent gene silencing and describe the kinetics and extent of chromatin modifications at the Oct4 locus in fibroblasts and pluripotent cells. H3K9me3 propagated symmetrically and continuously at average rates of ∼0.18 nucleosomes/hr to produce domains of up to 10 kb. After removal of the HP1α stimulus, heterochromatic domains were heritably transmitted, undiminished through multiple cell generations. Our data enabled quantitative modeling of reaction kinetics, which revealed that dynamic competition between histone marking and turnover, determines the boundaries and stability of H3K9me3 domains. This framework predicts the steady-state dynamics and spatial features of the majority of euchromatic H3K9me3 domains over the genome.
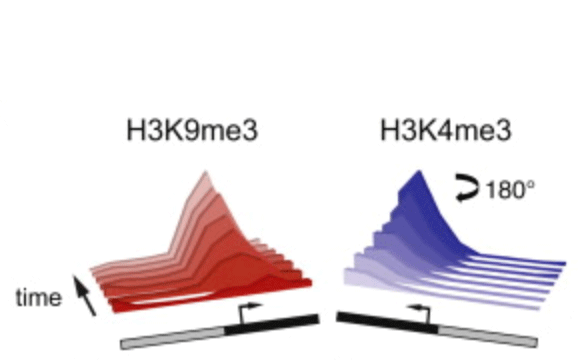
Hathaway NA*, Bell O*, Hodges C, Miller EL, Neel DS, and Crabtree GR. (2012) "Dynamics and memory of heterochromatin in living cells," Cell 149(7): 1447-14460.
2011
Atomic force microscopy of RNA polymerase transcribing through a nucleosome
Upon transcription, histones can either detach from DNA or transfer behind the polymerase through a process believed to involve template looping. The details governing nucleosomal fate during transcription are not well understood. Our atomic force microscopy images of yeast RNA polymerase II-nucleosome complexes confirm the presence of looped transcriptional intermediates and provide mechanistic insight into the histone-transfer process through the distribution of transcribed nucleosome positions. Notably, we find that a fraction of the transcribed nucleosomes are remodeled to hexasomes, and this fraction depends on the transcription elongation rate. A simple model involving the kinetic competition between transcription elongation, histone transfer and histone-histone dissociation quantitatively explains our observations and unifies them with results obtained from other polymerases. Factors affecting the relative magnitude of these processes provide the physical basis for nucleosomal fate during transcription and, therefore, for the regulation of gene expression.
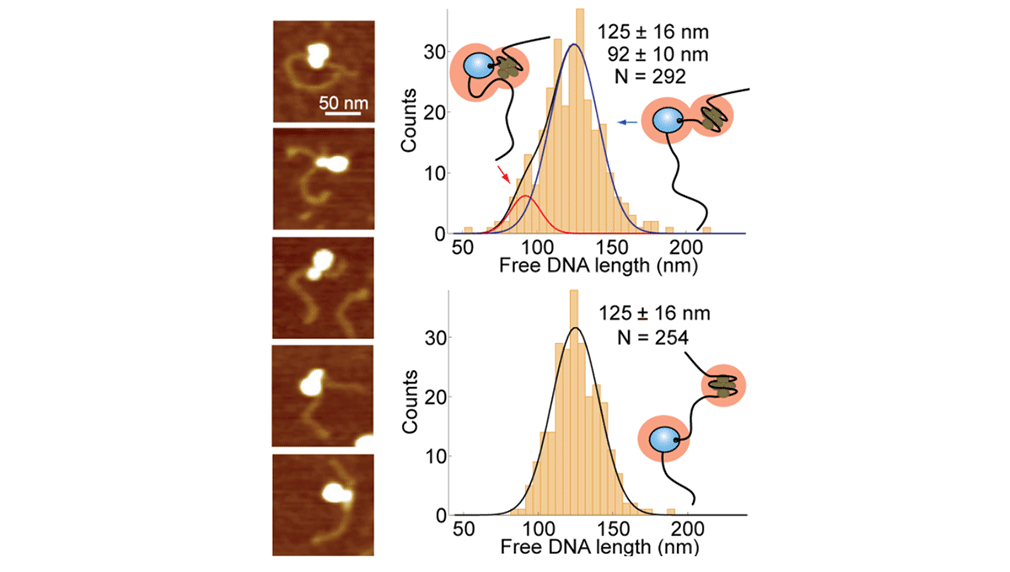
Bintu L*, Kopaczynska M*, Hodges C, Lubkowska L, Kashlev M, Bustamante C (2011) "The elongation rate of RNA polymerase determines the fate of transcribed nucleosomes," Nature Structural and Molecular Biology 18(12): 1394-1399.
Exertion of mechanical force during targeted protein degradation
AAA+ unfoldases denature and translocate polypeptides into associated peptidases. We report direct observations of mechanical, force-induced protein unfolding by the ClpX unfoldase from E. coli, alone, and in complex with the ClpP peptidase. ClpX hydrolyzes ATP to generate mechanical force and translocate polypeptides through its central pore. Threading is interrupted by pauses that are found to be off the main translocation pathway. ClpX's translocation velocity is force dependent, reaching a maximum of 80 aa/s near-zero force and vanishing at around 20 pN. ClpX takes 1, 2, or 3 nm steps, suggesting a fundamental step-size of 1 nm and a certain degree of intersubunit coordination. When ClpX encounters a folded protein, it either overcomes this mechanical barrier or slips on the polypeptide before making another unfolding attempt. Binding of ClpP decreases the slip probability and enhances the unfolding efficiency of ClpX. Under the action of ClpXP, GFP unravels cooperatively via a transient intermediate.
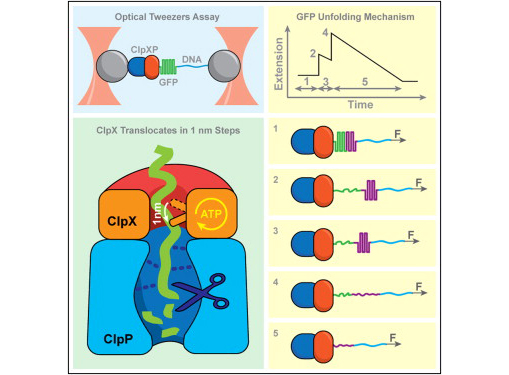
Maillard RA, Chistol G, Sen M, Righini M, Tan J, Kaiser CM, Hodges C, Martin A, Bustamante C (2011) "ClpX(P) generates mechanical force to unfold and translocate its protein substrates," Cell 145(3): 459-469.
2009
Observing individual transcription events through a nucleosome in real time
RNA polymerase II (Pol II) must overcome the barriers imposed by nucleosomes during transcription elongation. We have developed an optical tweezers assay to follow individual Pol II complexes as they transcribe nucleosomal DNA. Our results indicate that the nucleosome behaves as a fluctuating barrier that locally increases pause density, slows pause recovery, and reduces the apparent pause-free velocity of Pol II. The polymerase, rather than actively separating DNA from histones, functions instead as a ratchet that rectifies nucleosomal fluctuations. We also obtained direct evidence that transcription through a nucleosome involves transfer of the core histones behind the transcribing polymerase via a transient DNA loop. The interplay between polymerase dynamics and nucleosome fluctuations provides a physical basis for the regulation of eukaryotic transcription.
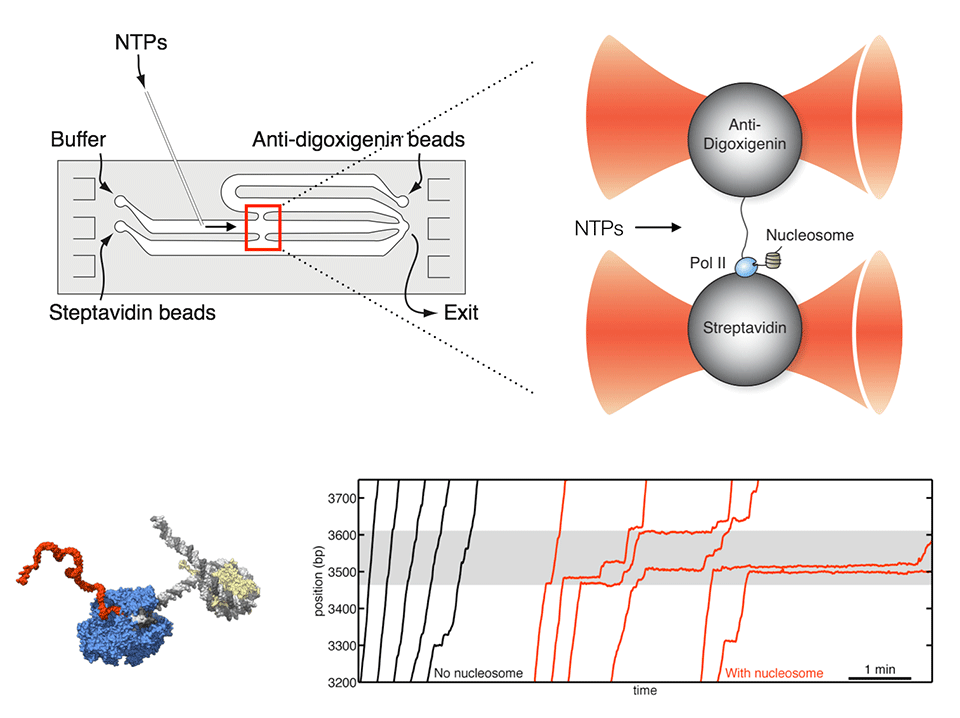
Hodges C*, Bintu L*, Lubkowska L, Kashlev M, and Bustamante C (2009) "Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II," Science 325(5940): 626-628.
2008
First observation of single translation events occurring in real time
We have followed individual ribosomes as they translate single messenger RNA hairpins tethered by the ends to optical tweezers. Here we reveal that translation occurs through successive translocation--and-pause cycles. The distribution of pause lengths, with a median of 2.8 s, indicates that at least two rate-determining processes control each pause. Each translocation step measures three bases--one codon-and occurs in less than 0.1 s. Analysis of the times required for translocation reveals, surprisingly, that there are three substeps in each step. Pause lengths, and thus the overall rate of translation, depend on the secondary structure of the mRNA; the applied force destabilizes secondary structure and decreases pause durations, but does not affect translocation times. Translocation and RNA unwinding are strictly coupled ribosomal functions.

Wen JD, Lancaster L, Hodges C, Zeri AC, Yoshimura SH, Noller HF, Bustamante C, Tinoco I (2008). "Following translation by single ribosomes one codon at a time," Nature 452(7187): 598-603.